Leverage the hemostatic potential of HMW multimers
The size of VWF multimers directly correlates with their in vitro function.
- High molecular weight (HMW) VWF multimers are more effective in supporting primary homeostasis, whereas low molecular weight (LMW) multimers are less functionally active1
- HMW-VWF multimers have been shown to be associated with increased hemostatic activity2
- HMW-VWF multimers are associated with shortened bleeding time2
The process of blood coagulation has two distinct stages of hemostasis: primary hemostasis and secondary hemostasis. While factor VIII (FVIII) has a role in secondary hemostasis, von Willebrand factor protein is a key component in primary hemostasis.3

People with von Willebrand disease (VWD) have an insufficient amount of VWF protein in the blood, or VWF that is defective. Because VWF is essential in primary hemostasis, an insufficient amount or a supply of defective VWF interferes with proper platelet adherence or aggregation.4 The mainstay of VWD treatment is the replacement of the deficient VWF protein. This results in shortened bleeding time and correction of the coagulation abnormality.5
The VWF profile of HUMATE-P correlates to hemostatic activities during the stages of hemostasis
HMW-VWF multimers promote6:
- Effective collagen binding
- Effective platelet adhesion
- Effective platelet aggregation
As close as replacement therapy multimer patterns get to normal human plasma with appropriate FVIII levels6
Like normal human plasma, HUMATE-P contains a high percentage of HMW-VWF multimers, and can correct the hemostatic defect in patients with VWD.6
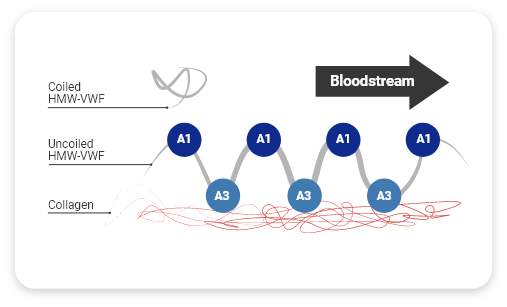
HMW-VWF multimers promote effective collagen-binding
When the vessel is injured, several actions occur to form the initial hemostatic plug1:
- VWF unfolds from its inactivated pretzel-like shape, exposing A1 domains (platelet receptors)
- Collagen binds with A3 domains (collagen-binding sites)
HMW-VWF multimers have more A3 domains and A1 domains than LMW-VWF multimers, allowing more bonds to form.1
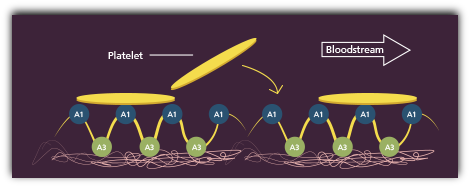
HMW-VWF multimers promote effective platelet adhesion
At the same time, platelets rush to the injury site, forming bonds with A1 domains.1
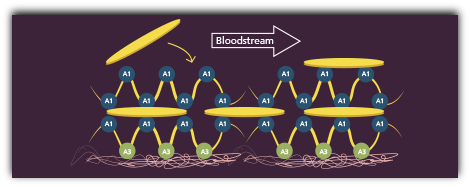
HMW-VWF multimers promote effective platelet aggregation
The platelet bridges and VWF form layers, known as the initial hemostatic plug, which stops the bleeding at the injury site.1
Successful completion of both stages of hemostasis results in the final step of the clotting process, the formation of a stable hemostatic plug.
HUMATE-P has 94% homology with normal human plasma (NHP).2
References
- Reininger AJ. Function of von Willebrand factor in haemostasis and thrombosis. Haemophilia. 2008;14(Suppl 5):11-26.
- Metzner HJ, Hermentin P, Cuesta-Linker T, Langner S, Müller H-G, Friedebold J. Characterization of factor VIII/von Willebrand factor concentrates using a modified method of von Willebrand factor multimer analysis. Haemophilia. 1998;4(Suppl 3):25-32.
- Stockschlaeder M, Schneppenheim R, Budde U. Update on von Willebrand factor multimers: focus on high-molecular-weight multimers and their role in hemostasis. Blood Coagul Fibrinolysis. 2013;25:206-216.
- The Diagnosis, Evaluation, and Management of von Willebrand Disease. US Department of Health and Human Services. National Institutes of Health. National Heart, Lung, and Blood Institute. NIH Publication No. 08-5832. Washington, DC. December 2007.
- Peyvandi F, Kouides P, Turecek PL, Dow E, Berntorp E. Evolution of replacement therapy for von Willebrand disease: from plasma fraction to recombinant von Willebrand factor. Blood Reviews. 2019;38:10057. https://doi.org/10.1016/j.blre.2019.04.001. Accessed October 6, 2020.
- Budde U, Metzner HJ, Müller H-G. Comparative analysis and classification of von Willebrand Factor/Factor VIII concentrates: impact on treatment of patients with von Willebrand disease. Semin Thromb Hemost. 2006;32:626-635.